На прошедшей в Кембриджском университете конференции «Cell Senescence in Cancer and Ageing» было дано такое определение клеточного старения: «Клеточным старением называется устойчивая остановка пролиферации, вызванная различными молекулярными триггерами, включающими активацию онкогенов, а также избыточное количество клеточных делений. Кроме того, сенесцентные клетки характеризуются секрецией целого ряда стромальных регуляторов и регуляторов воспаления (так называемым «ассоциированным со старением сереторным фенотипом»), влияющих на функционирование соседних клеток, включая иммунокомпетентные. Целый ряд убедительных фактов свидетельствует о том, что клеточное старение представляет собой эффективный механизм подавления опухолевого роста. В тоже время, клеточное старение возможно вносит свой вклад в старение тканей и всего организма».
Из-за разных причинных механизмов, выделяется три вида клеточного старения.
Самым первым в начале 60-х годов прошлого века было обнаружено клеточное репликативное старение. В ставшей уже знаменитой работе американские геронтологи Л. Хейфлик и П. Мурхед в экспериментах с культивируемыми человеческими фибробластами установили, что клетки не делятся бесконечно и есть предел клеточному делению (названный впоследствии пределом, или лимитом Хейфлика) [1]. Спустя 10 лет советский биолог Алексей Оловников дал логическое объяснение этому феномену, связав лимит клеточных делений с постепенным укорочением концевых участков ДНК, теломер. Связано это с тем, что фермент теломераза, способный наращивать теломеры после их укорочения, не активен в большинстве соматических клеток. После того, как теломеры укорачиваются до критического уровня, возникает ответ на повреждение ДНК (DNA damage response, DDR), в результате чего происходит остановка клеточного цикла и клетка переходит в разряд сенесцентных. Известно, что внешние факторы, которые отрицательно влияют на здоровье и долголетие (ожирение, отсутствие физических упражнений, стресс), также оказывают негативное влияние и на укорочение теломер [2]. Также ускорение сокращения длины теломер наблюдается у пациентов с нейродегенеративными заболеваниями, такими как болезнь Альцгеймера [3].
Считается, что для большинства клеток лимит Хейфлика составляет около 50 делений, после чего клетка перестаёт делиться. Чтобы отличать старение организма в целом от клеточного старения, Хейфлик и Мурхед ввели в научный оборот специальный термин, обозначающий старение клеток – senescence (в отличии от старения организма – aging).
В дополнение к репликативному старению, старение клеток может быть вызвано также и другими факторами, которые преждевременно индуцируют клеточное старение независимо от длины теломер. Эти факторы составляют второй и третий типы клеточного старения.
Так, активация онкогенов, таких как RAS и RAF, вызывает клеточное старение, названное онкоген-индуцированным клеточным старением (oncogene-induced senescence, OIS). Эта форма клеточного старения связана с подавлением опухоли. Геномные сравнительные исследования клеток с репликативным и OIS старением показывают, что, хотя существуют некоторые общие изменения экспрессии генов между этими двумя видами по сравнению с пролиферирующими клетками, также имеются и существенные различия [4]. Известно, что в механизмах OIS-старения большую роль играют повреждения ДНК, связанные с активными формами кислорода (АФК). Также в возникновении OIS активно участвует ERK-киназа, стимулируя деградацию белков, необходимых для прогрессирования клеточного цикла. Роль ответа на повреждение ДНК (DDR) в этом виде клеточного старения не донца выяснена. Известно, что мутантные онкогены, такие, как H — Ras G12V, обладают потенциалом для активации молекулярных путей клеточного старения, связанных с киназой р38 MAPK и транскрипционным фактором NF-кВ, независимо от повреждения ДНК. Онкогенный ген Ras также может способствовать повышению регуляции р53 через p19ARF и клеточному старению независимо от повреждения ДНК. [5]. Поэтому не исключается стимуляция клеточного OIS старения даже в отсутствие повреждения ДНК.
Третьим видом клеточного старения, также не зависящим от длины теломер, является стресс-индуцированное преждевременное клеточное старение (stress-induced premature senescence, SIPS). Оно возникает в ответ на стрессовые факторы различной природы: ионизирующее и ультрафиолетовое излучение, увеличение уровня АФК, химиотерапевтические препараты. В отличии от OIS-старения возникновение SIPS полностью зависит от ответа на повреждение ДНК (DDR). Фенотипически SIPS и репликативное клеточное старение во многом сходны, но могут различаться на уровне экспрессии белка. Роль SIPS в общем старении организма остаётся не до конца ясной – повышенная экспрессия антиоксидантов и подавление АФК, основных факторов возникновения SIPS, не приводило к увеличению срока жизни [6].
Молекулярные механизмы остановки клеточного цикла в сенесцентных клетках сегодня активно изучаются. Известно, что степень повреждения ДНК по-разному влияет на клеточный цикл. Так, умеренное повреждение ДНК может индуцировать временную остановку роста, обширное повреждение ДНК вызывает запрограммированную гибель клеток, стойкое повреждение ДНК вызывает старение клеток. Молекулярные детерминанты (основные факторы), которые регулируют переход от временной приостановки роста к необратимому аресту цикла, сложны и еще не полностью описаны. Известно, что повреждения ДНК первоначально активируют путь р53- р21, который останавливает клеточный цикл. Затем, если повреждения ДНК не репарируются, клетка или уходит в апопотоз, или становится сенесцентной. Во втором случае ключевую роль играет белок p16 INK4a, который регулирует долгосрочное сохранение остановки клеточного цикла через сигнальный путь рRb-E2F (белок ретинобластомы, рRb –транскрипционный фактор E2F) и изменения структуры хроматина [7].
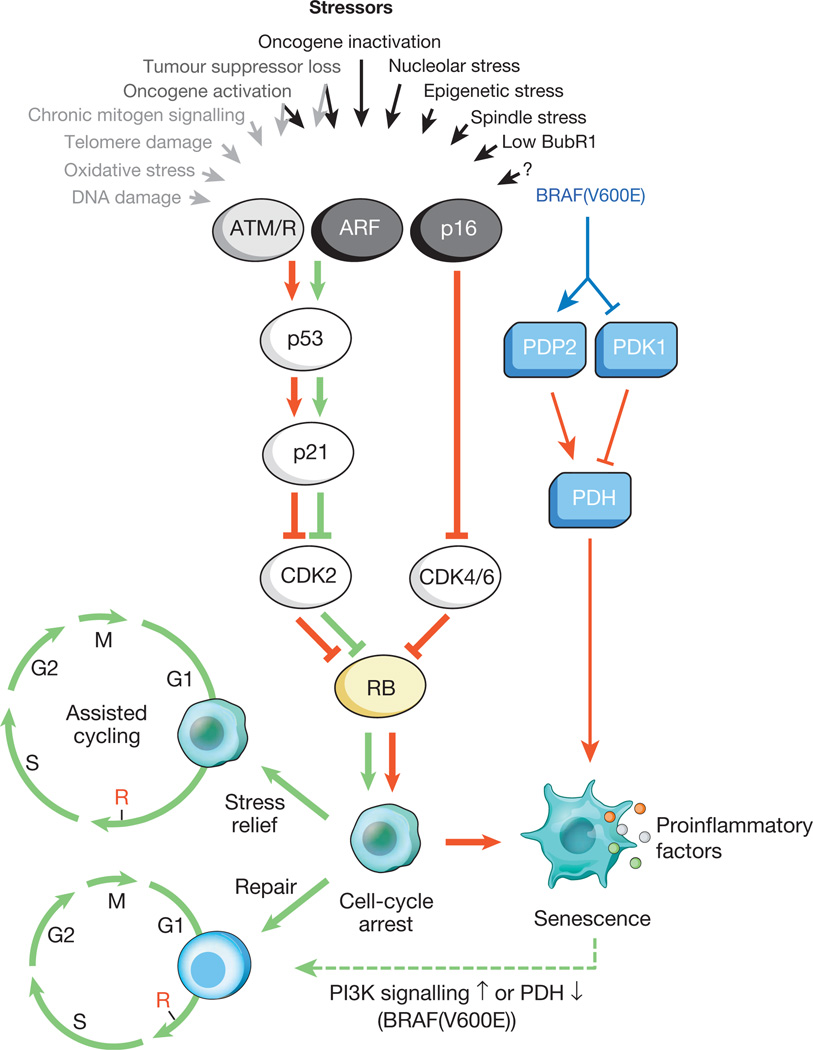
Рис.1. Стимулы, вызывающие клеточное старение, и основные эффекторные пути
Разнообразные внутриклеточные и внешние стрессы могут активировать программу клеточного старения. Эти стрессоры захватывают различные клеточные сигнальные каскады, и в итоге активируют p53 и p16 INK4a. Типы стресса, которые активируют передачу сигналов p53 через DDR, обозначены серым текстом и стрелками (ROS (АФК) вызывают ответ на повреждение ДНК (DDR), нарушая транскрипцию гена и репликацию ДНК, а также сокращая теломеры). Активированный p53 индуцирует p21, который вызывает временную остановку клеточного цикла путем ингибирования циклина E-Cdk2. p16 INK4a также ингибирует прогрессирование клеточного цикла, но делает это, нацеливаясь на комплексы циклин D-Cdk4 и циклин D-Cdk6. Оба p21 и p16 INK4a действуют, предотвращая инактивацию Rb, что приводит к продолжению репрессии генов-мишеней E2F, необходимых для начала S-фазы. При сильном стрессе (красные стрелки) временно блокированные клетки переходят в стадию ареста клеточного цикла. Клетки, подверженные незначительному повреждению, могут быть успешно восстановлены и возобновить нормальный цикл. Таким образом, путь p53-p21 может либо антагонизировать, либо синергизировать своё действие с p16 INK4a в пожилом возрасте в зависимости от типа и уровня стресса. BRAF (V600E) связан со старением через метаболический эффекторный путь. BRAF (V600E) активирует PDH, индуцируя PDP2 и ингибируя экспрессию PDK1, способствуя сдвигу от гликолиза к окислительному фосфорилированию, которое создает вызывающий старение окислительно-восстановительный стресс. Клетки, подвергающиеся старению, индуцируют воспалительный транскриптом независимо от связанного со старением индуцирующего стресса (цветные точки представляют различные факторы SASP). Красные и зеленые стрелки, соответственно, указывают на активность, способствующую старению и «предотвращению старения». Штриховая зеленая стрелка обозначает механизм «смены старения».
Известно, что сенесцентные клетки активно влияют на своё микроокружение (окружающие их ткани), секретируя целый ряд активных молекул: провоспалительные цитокины, хемокины факторы роста, протеазы (всего около 40 различных видов молекул). Эти вещества были сведены в единую группу – ассоциированный с клеточным старением секреторный фенотип (senescence associated secretory phenotype, SASP). Известно, что факторы SASP активно участвуют в ремоделировании тканей в эмбриональном развитии. Причём под их воздействием перестраивается как организм матери, так и эмбриона. Предполагается, что эволюционная природа SASP связана с рядом защитных механизмов: подавления опухолей, восстановления после травм и регенерацией тканей.
Упрощённо физиологическое действие SASP можно описать следующим образом. Секретируемые провоспалительные молекулы формируют вокруг подлежащих удалению сенесцентных клеток очаг воспаления. Что привлекает в данное место клетки иммунной системы для элиминации стареющих клеток. Входящие в SASP матриксные металлопротеазы (ММР-1, ММР-10, ММР-3) и сериновые протеазы ремоделируют внеклеточный матрикс, чтобы облегчить проникновение клеток иммунной системы к стареющим клеткам. И, наконец, секретируемые ростовые факторы стимулируют размножение соседних клеток для замещения удалённых сенесцентных.
Это описано так, как механизм SASP должен работать в норме, в молодом и здоровом организме. Но с возрастом и при отклонениях его эффективность может существенно снижаться, что вызывает накопление сенесцентных клеток в тканях и, как следствие – к продолжительной секреции провоспалительных факторов SASP. Что сопровождается возникновением очагов хронического воспаления. Кроме этого, известно, что продолжительная активность SASP действует как инфекция на нормальные клетки. Секретируемые сенесцентными клетками активные молекулы попадают во внеклеточное пространство и, воздействуя на соседние нормальные клетки, инициируют арест клеточного цикла и остановку пролиферации. Что в значительной степени ускоряет развитие клеточного старения в тканях.
Кроме этого, длительная секреция стареющими клетками факторов SASP связана с развитием возрастных патологий. Так, повышенная секреция матриксных металлопротеаз сенесцентными клетками стимулирует развитие ишемической болезни сердца, остеопороза и остеоартрита. Сенесцентные гладкомышечные клетки участвуют в развитии атеросклероза, посредством секреции большого количества провоспалительных цитокинов. Продолжительная секреция провоспалительного фактора TNF-α сенесцентными Т-клетками участвует в процессах, связанных с дисфункцией костной ткани. Кроме этого известно, что повышение уровня провоспалительного IL-6 связано с резистентностью к инсулину, диабетом, атеросклерозом и болезнями печени. Для обозначения всех этих процессов, связанных с системным хроническим воспалением и старением, в которых ключевую роль играют именно факторы SASP, был введён специальный термин – inflammaging. Кроме этого, была описана двойная роль SASP в канцерогенезе – его опухолесупрессорная и опухолепромотирующая активность [8].
В связи с описанной выше двойной ролью клеточного старения в молодом и пожилом возрасте некоторые учёные рассматривают клеточное старение как пример эволюционной антагонистической плейотропии, которую ещё шутливо формулируют фразой «пользуйся сейчас – заплатишь потом». Согласно этой теории, процессы, которые закрепились в эволюции для повышения выживаемости молодых организмов, могут иметь накапливающиеся вредные эффекты у более старых особей. Так, механизмы, связанные с сенесцентными клетками, в молодом возрасте задействованы при беременности, восстановлении после травм, защите от опухолей. В более же пожилом возрасте те же самые механизмы вызывают системное воспаление, дегенерацию тканей и развитие патологий [9].
Авторы концепции SENS среди пула стареющих клеток организма особо выделяют два вида, связанные с жировой тканью: преадипоциты и клетки висцеральной жировой ткани. И это не случайно, так как сегодня известно, что висцеральный жир является метаболически активным и высвобождает целый ряд активных молекул – адипокинов. Которые, в свою очередь, связаны с развитием целого ряда возрастных патологий (резистентностью к инсулину, диабетом, сердечно-сосудистыми болезнями). Также авторы SENS акцентируют внимание на возрастном ухудшении работы иммунной системы. По их мнению, это связано с перегрузкой организма стареющими клетками и, как ответ – к перепроизводству уничтожающих сенесцентные клетки Т-киллеров в ущерб другим видам иммунных клеток. Что делает стареющий организм уязвимым со стороны различных инфекций.
Решение проблемы накопления сенесцентных клеток авторы SENS видят в двух направлениях. Первое – разработка препаратов, токсичных для старых клеток, или вызывающих их апоптоз, но при этом безвредных для здоровых, нормальных клеток. И второе направление – поиск стимуляторов иммунной системы, чтобы выборочно искать и убивать стареющие клетки-мишени. Наиболее вероятным способом выборочной атаки на эти аномальные клетки, по мнению разработчиков SENS, было бы использование отличительных молекул, которые встречаются на их поверхностях. Действительно, разные типы клеток имеют различия своих поверхностей. Поэтому первым шагом стоит вопрос идентификации и ориентации маркеров поверхности клеток, которые являются специфическими для подлежащих удалению сенесцентных клеток. Эта стратегия не абстрактна, а уже является основой некоторых методов лечения рака, которые блокируют рост клеток и привлекают иммунные клетки для их уничтожения [10].
Проведённые исследования показали, что сенесцентные клетки действительно накапливаются с возрастом в различных тканях [11]. Также было описано, что устранение стареющих клеток, которые накапливаются в модели ускоренно стареющих мышей, предотвращает появление трех основных фенотипов старения (катаракта, саркопения, потеря подкожного жира) [12]. В связи с этим, вполне очевидно возникает необходимость в идентификации надёжных и эффективных биомаркеров клеточного старения. Которые необходимы в первую очередь, для отслеживания действия потенциальных препаратов-сенолитиков.
Наиболее часто в качестве биомаркера сенесцентных клеток используется ассоциированная с клеточным старением β-галактозидаза (senescence-associated beta-galactosidase, SA-β-Gal). Фермент β-галактозидаза – это лизосомная гидролаза, которая расщепляет концевую бета-галактозу из содержащих ее соединений (лактозы, кератинсульфатов, сфинголипидов и др.). Ещё в 1995 году было описано, что экспрессия SA-β-Gal значительно возрастает в сенесцентных клетках. Для определения её содержания в стареющих тканях используется иммуногистохимический метод. В качестве альтернативного метода определения активности SA-β-gal в клетках применяется проточная цитометрия с использованием 5-додеканоиламинофлуоресцеина ди-β-D-галактопиранозида в качестве субстрата.
Однако применение SA-β-Gal в качестве биомаркера клеточного старения имеет свои ограничения, так как этот фермент может давать ложные срабатывания, повышая экспрессию не только в стареющих клетках, но и в «молодых», у которых по разным причинам происходит ограничение пролиферации. Поэтому сегодня считается целесообразным использовать SA-β-Gal вместе с другими маркерами клеточного старения.
В 2017 году израильские цитологии разработали новую, более эффективную технологию на основе проточной цитометрии, с использованием цитометра ImageStreamX. Этот метод позволял обнаруживать SA-β-Gal в тканях с эффективностью более 80 %. Для повышения эффективности анализа учёные вместе с SA-β-Gal определяли ещё несколько биомаркеров клеточного старения – белки HMGB1 и γH2AX. HMGB1 – белок из группы ядерных негистоновых белков HMG, в сенесцентных клетках он покидает ядро и перемещается во внеклеточное пространство. γH2AX – фосфорилированная форма гистона H2AX, является признанным маркером раннего повреждения ДНК и клеточного старения. Кроме этого, новый метод израильских учёных позволял определять стареющие клетки по их увеличенным размерам. По мнению исследователей, их технология может быть использована для быстрого и определения эффективности новых фармацевтических соединений, которые будут специально разработаны для устранения стареющих клеток из тканей. [13].
Ещё одним биомаркером старения могут выступать ассоциированные с клеточным старением гетерохроматиновые фокусы (SAHF). SAHF – это особые гетерохроматиновые структуры, которые образуются в ядрах стареющих клеток. Их образование связано с необратимой геторохроматинизацией, связанной с инактивацией расположенных на этом участке генов, участвующих в клеточном цикле (MCM3, PCNA, Cyclin A). SAHF можно увидеть под микроскопом после их окрашивания специальным красителем DAPI. Кроме этого, описана повышенная экспрессия промиелоцитарного лейкозного белка (PML) в сенесцентных клетках, который также может быть дополнительным маркером клеточного старения[14].
Американские исследователи описали в качестве биомаркера клеточного старения белок p16 INK4a из Т-клеток периферической крови человека. p16 INK4a как уже было описано выше, принимает самое активное участие в остановке клеточного цикла сенесцентных клеток. Экспрессия p16 INK4a повышается в сенесцентных клетках и, как оказалось, была значительно связана с курением и физической неактивностью. Кроме того, экспрессияp16 INK4a была связана с концентрацией плазменного IL-6, маркером возрастного воспаления. По мнению учёных, экспрессия p16 INK4a представляет собой легко измеряемый биомаркер периферической крови для определения клеточного старения [15].
Приложение 1.
Биомаркеры клеточного старения.
1. Ассоциированная с клеточным старением β-галактозидаза (senescence-associated beta-galactosidase, SA-β-Gal).
2. Белок HMGB1 (high-mobility group protein B1).
3. Фосфорилированный гистон γH2AX.
4. Ассоциированные с клеточным старением гетерохроматиновые фокусы (SAHF).
5. Белок промиелоцитарного лейкоза (PML).
6. Белок p16 INK4a.
2. Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD, Cawthon RM. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004 Dec 7;101(49):17312-5.
3. Panossian LA, Porter VR, Valenzuela HF, Zhu X, Reback E, Masterman D, Cummings JL, Effros RB. Telomere shortening in T cells correlates with Alzheimer's disease status. Neurobiol Aging. 2003 Jan-Feb;24(1):77-84.
4. Nelson DM1, McBryan T, Jeyapalan JC, Sedivy JM, Adams PD. A comparison of oncogene-induced senescence and replicative senescence: implications for tumor suppression and aging. Age (Dordr). 2014 Jun;36(3):9637.
5. Palmero I, Pantoja C, Serrano M. p19ARF links the tumour suppressor p53 to Ras. Nature. 1998 Sep 10; 395(6698):125-6.
6. Pérez VI1, Van Remmen H, Bokov A, Epstein CJ, Vijg J, Richardson A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell. 2009 Feb;8(1):73-5.
7. Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003 Jun 13; 113(6):703-16.
8. Бородкина А.В., Дерябин П.И., Грюкова А.А., Никольский Н.Н. «Социальная жизнь» стареющих клеток: что такое SASP и зачем его изучать? Acta Naturae, 2018, 10(1). С.4-15.
9. Shankar J. Chinta, Georgia Woods, Anand Rane, Marco Demaria, Judith Campisi, and Julie K Andersen. Cellular senescence and the aging brain. Exp Gerontol. 2015 Aug; 68: 3–7.
10. ApoptoSENS: Removing dysfunctional cells.
11. Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev. 2007 Jan;128(1):36-44.
12. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011 Nov 2;479(7372):232-6.
13. Anat Biran, Lior Zada, Paula Abou Karam, Ezra Vadai, Lior Roitman, Yossi Ovadya, Ziv Porat, and Valery Krizhanovsky. Quantitative identification of senescent cells in aging and disease. Aging Cell. 2017 Aug; 16(4): 661–671.
14. Bruno Bernardes de Jesus and Maria A. Blasco. Assessing Cell and Organ Senescence Biomarkers. Circ Res. 2012 Jun 22; 111(1): 97–109.
15. Yan Liu, Hanna K. Sanoff, Hyunsoon Cho, Christin E. Burd, Chad Torrice, Joseph G Ibrahim, Nancy E. Thomas, and Norman E. Sharpless. Expression of p16INK4a in peripheral blood T-cells is a biomarker of human aging. Aging Cell. 2009 Aug; 8(4): 439–448.
Комментариев нет:
Отправить комментарий